经常有朋友问起自己要做什么分析,推荐一个电脑的配置。通常限制程序运行的最主要因素是内存,内存不足程序会直接运行不起来,CPU性能弱顶多是运行的慢,硬盘比较便宜,不需要特别评估。
针对这个问题,我们准备推出一系列测试推文,统计计算常用软件的运行时间、所需的最大物理内存 (后面统计的都是所需最大物理内存,也就是配电脑所需的内存;程序运行过程中可能还会使用虚拟内存,这个参数也有统计,但不在分析范围之内)、生成文件所占的磁盘空间,以给后续选择做参考依据。
资源统计主要基于Time除了监控程序运行时间还能干这个?提到的time函数。
测试 STAR 构建基因组索引所需的计算资源
基因组序列和基因注释文件下载和整理
# 下载当前最新版本的基因组 wget -c http://ftp.ensembl.org/pub/release-104/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz # 处理基因组序列文件,染色体名字增加 chr # 且名字简化,去除无关信息 gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz -c | sed 's/^>/>chr/' | cut -d ' ' -f 1 > GRCh38.fa # 下载当前最新基因注释文件 wget -c http://ftp.ensembl.org/pub/release-104/gtf/homo_sapiens/Homo_sapiens.GRCh38.104.gtf.gz # 给染色体名字增加 chr gunzip Homo_sapiens.GRCh38.104.gtf.gz -c | grep -v '^#' | sed 's/^/chr/' >GRCh38.gtf
测试构建完整人类基因组所需的资源
调用 STAR 程序构建基因组索引
/usr/bin/time -o STAR_human_genome.log -v STAR --runMode genomeGenerate --runThreadN 2 --genomeDir star_GRCh38 --genomeFastaFiles GRCh38.fa --sjdbGTFfile GRCh38.gtf > STAR_human_genome_run.log 2>&1
统计信息输出如下(这里只关注下面的 3 个统计信息,CPU利用率、运行耗时、最大物理内存):
Percent of CPU this job got: 174% Elapsed (wall clock) time (h:mm:ss or m:ss): 1:36:08 Maximum resident set size (kbytes): 36945552
将人类基因组按染色体拆分模拟不同大小基因组构建索引的计算资源需求
采用染色体累加的方式,不断模拟不同大小的基因组对计算资源的需求。
/bin/rm -f GRCh38_tmp.seqid
for i in `seq 1 22`; do
# 累加染色体,第一次循环测试 chr1,
# 第二次循环测试 chr1,chr2
echo "chr$i" >>GRCh38_tmp.seqid
# 调用 seqkit 提取序列
seqkit grep -f GRCh38_tmp.seqid GRCh38.fa -o GRCh38_tmp.fa
# 提取对应的 gtf 注释
awk 'BEGIN{OFS=FS="\t"}ARGIND==1{id[$1]=1}ARGIND==2{if(id[$1]==1) print $0}' GRCh38_tmp.seqid GRCh38.gtf >GRCh38_tmp.gtf
# 统计构建基因组索引所需的计算资源
/usr/bin/time -o STAR_human_genome_${i}.log -v STAR --runMode genomeGenerate --runThreadN 2 --genomeDir star_GRCh38_${i} --genomeFastaFiles GRCh38_tmp.fa --sjdbGTFfile GRCh38_tmp.gtf > STAR_human_genome_${i}.run_log 2>&1
done
统计输出结果同上,比如对chr1构成的基因组统计结果如下:
Percent of CPU this job got: 91% Elapsed (wall clock) time (h:mm:ss or m:ss): 5:33.95 Maximum resident set size (kbytes): 7719688
整合数据生成最终测试结果数据集
首先写一个awk脚本,整理并转换 CPU 使用率、程序耗时、最大物理内存需求。这个脚本存储为timeIntegrate.awk。
#!/usr/bin/awk -f
# 定义一个世界转换函数
function to_minutes(time_str) {
a=split(time_str,array1,":");
minutes=0;
count=1;
for(i=a;i>=1;--i) {
minutes+=array1[count]*60^(i-2);
count+=1;
}
return minutes;
}
# 设定特殊的输入分隔符,注意冒号后面的空格
BEGIN{OFS="\t"; FS=": "; }
# 数据量计量
ARGIND==1{datasize=$0;}
# 解析time的输出
ARGIND==2{
if(FNR==1 && outputHeader==1)
print "Time_cost\tMemory_cost\tnCPU\tData_size";
if($1~/Elapsed/) {
# 时间转为分钟
time_cost=to_minutes($2);
} else if($1~/Maximum resident set size/){
# 内存转为 G
memory_cost=$2/10^6;
} else if($1~/CPU/) {
# cpu 计算为 n核
cpu=($2+0)/100};
}
END{print time_cost, memory_cost, cpu, datasize}
使用上面的脚本,整合所有基因组数据构建所需的计算和时间资源信息。
/bin/rm -f GRCh38_star_genome_build.summary
for i in `seq 1 22`; do
awk -v i=${i} 'BEGIN{for(j=1;j<=i;j++) scaffold["chr"j]=1;}\
{if(scaffold[$1]==1) genomeSize+=$2}END{print genomeSize/10^9}' star_GRCh38/chrNameLength.txt | \
awk -v outputHeader=$i -f ./timeIntegrate.awk - STAR_human_genome_${i}.log >>GRCh38_star_genome_build.summary
done
awk '{genomeSize+=$2}END{print genomeSize/10^9}' star_GRCh38/chrNameLength.txt \
| awk -f ./timeIntegrate.awk - STAR_human_genome.log >>GRCh38_star_genome_build.summary
最终获得结果文件如下:
Time_cost Memory_cost nCPU Data_size 5.56583 7.71969 0.91 0.248956 11.6128 15.7351 0.93 0.49115 14.7547 18.7495 1.09 0.689446 15.9043 24.3767 1.34 0.87966 23.3608 30.4486 1.64 1.0612 28.578 35.4594 1.49 1.232 33.4185 33.4571 1.41 1.39135 37.9005 32.8232 1.34 1.53649 39.8342 35.409 1.38 1.67488 39.5383 34.7446 1.5 1.80868 40.0458 34.5746 1.6 1.94377 42.4697 34.4593 1.61 2.07704 46.1803 33.432 1.54 2.19141 53.1342 33.9707 1.54 2.29845 56.1117 32.4766 1.51 2.40044 58.9417 31.6688 1.48 2.49078 59.9543 31.4797 1.51 2.57404 61.1 32.743 1.54 2.65441 69 32.921 1.64 2.71303 69.0333 33.692 1.65 2.77747 75.9 34.1749 1.7 2.82418 89.2667 34.7063 1.76 2.875 96.1333 36.9456 1.74 3.09975
部分构建好的索引因为硬盘不足删除了,后期才想着应该统计下,故做了手动添加:
Time_cost (minutes) Memory_cost (G) nCPU Data_size (G) Index_size (G) 5.56583 7.71969 0.91 0.248956 3.6 11.6128 15.7351 0.93 0.49115 NA 14.7547 18.7495 1.09 0.689446 7.5 15.9043 24.3767 1.34 0.87966 NA 23.3608 30.4486 1.64 1.0612 11 28.578 35.4594 1.49 1.232 NA 33.4185 33.4571 1.41 1.39135 14 37.9005 32.8232 1.34 1.53649 NA 39.8342 35.409 1.38 1.67488 16 39.5383 34.7446 1.5 1.80868 NA 40.0458 34.5746 1.6 1.94377 19 42.4697 34.4593 1.61 2.07704 NA 46.1803 33.432 1.54 2.19141 21 53.1342 33.9707 1.54 2.29845 NA 56.1117 32.4766 1.51 2.40044 23 58.9417 31.6688 1.48 2.49078 NA 59.9543 31.4797 1.51 2.57404 24 61.1 32.743 1.54 2.65441 NA 69 32.921 1.64 2.71303 25 69.0333 33.692 1.65 2.77747 NA 75.9 34.1749 1.7 2.82418 26 89.2667 34.7063 1.76 2.875 NA 96.1333 36.9456 1.74 3.09975 28
构建索引的时间随数据量的变化
构建基因组的索引所需时间跟基因组大小成正相关,大体分为 3 个阶段:
- 1.8 G以内基因组构建索引的时间与基因组大小近乎完美的正相关。
- 1.8 G - 2.3 G大小的基因组构建索引的时间基本相当,受基因组大小影响较小。
- 2.3 G - 3 G大小的基因组构建索引的时间与基因组大小正相关,且时间随基因组大小变化的幅度大于基因组大小在1.8 G以内时。
- 推测更大的基因组,时间需求也会更大。
此处只考虑了大小,但基因组序列组成的复杂性也会对构建索引有很大影响。
另外,后面应该尝试学习个模型做个简单预测。
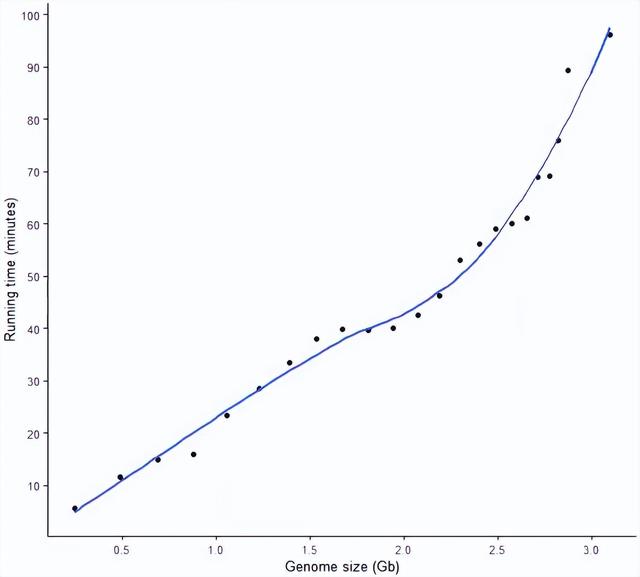
# devtools::install_git("https://gitee.com/ct586/ImageGP")
library(ImageGP)
library(ggplot2)
sp_scatterplot("GRCh38_star_genome_build.summary", melted = T, xvariable = "Data_size",
yvariable = "Time_cost", smooth_method = "auto",
x_label ="Genome size (Gb)", y_label = "Running time (minutes)") +
scale_x_continuous(breaks=seq(0.5,3, by=0.5)) +
scale_y_continuous(breaks=seq(10,100,length=10))
构建索引的所需内存随数据量的变化
- 在基因组大小超过1 G时,STAR 构建基因组索引需要的内存已经达到30 G。
- 基因组大小在1.2 G - 2.8 G时,STAR 构建基因组索引需要的内存变化不大,在31 G-35 G之间徘徊,甚至出现了基因组变大,而所需内存变少的现象。
- (还不知道怎么解释这一现象)
- 基因组大小在3 G时,需要的内存是37 G。
- 推测导致内存增加的不只是因为数据量的增加,更因为数据变得更复杂了(这部分增加的基因组大小来源于性染色体和不能确定来源的基因组区域)
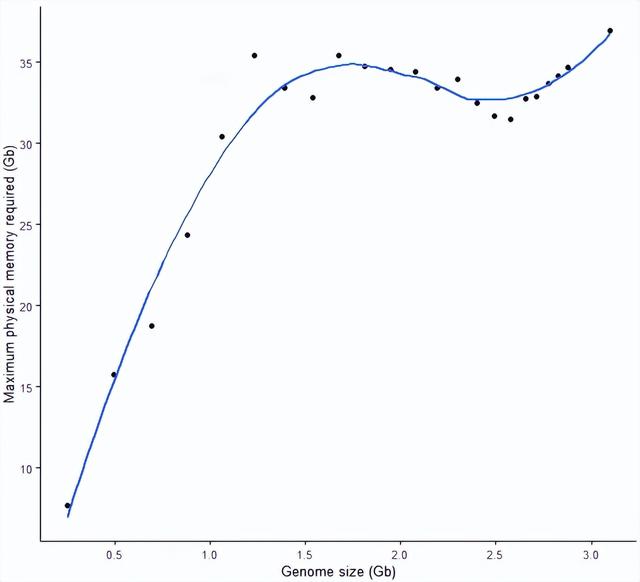
sp_scatterplot("GRCh38_star_genome_build.summary", melted = T, xvariable = "Data_size",
yvariable = "Memory_cost", smooth_method = "auto",
x_label ="Genome size (Gb)", y_label = "Maximum physical memory required (Gb)") +
scale_x_continuous(breaks=seq(0.5,3, by=0.5)) +
scale_y_continuous(breaks=seq(5,40, by=5))
构建索引时对 CPU 的利用率
在构建索引时,我们只给程序分配了 2 个线程;检测结果也有些出乎意料,STAR 似乎对多线程的利用并不充分,尤其时基因组比较小时,可能更多还是 IO 密集型的操作。
从趋势来看,随着基因组大小变大,CPU 的双线程利用率也是逐渐增高的。这也就是说,有时核也不需要给太多,没那么大数据量时,给了多核不过是心里安慰。
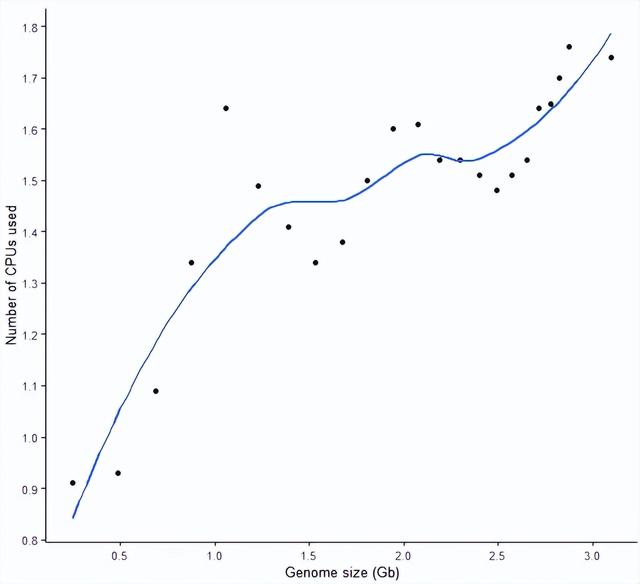
sp_scatterplot("GRCh38_star_genome_build.summary", melted = T, xvariable = "Data_size",
yvariable = "nCPU", smooth_method = "auto",
x_label ="Genome size (Gb)", y_label = "Number of CPUs used") +
scale_x_continuous(breaks=seq(0.5,3, by=0.5)) +
scale_y_continuous(breaks=seq(0,2, by=0.1))
生成的索引大小根基因组大小的关系 (磁盘需求)
最开始统计时忽略了这个信息,本来time是可以输出File system outputs信息的,但不知道在这怎么会都是0,可能是操作系统的问题。
简单的根据最终生成的索引大小来做个分析和判断,一个近乎完美的正相关,基因组越大,产生的索引越大,大约是8-9倍基因组的大小。一个完整的人类基因构建的 STAR 索引需要28 G的硬盘空间。
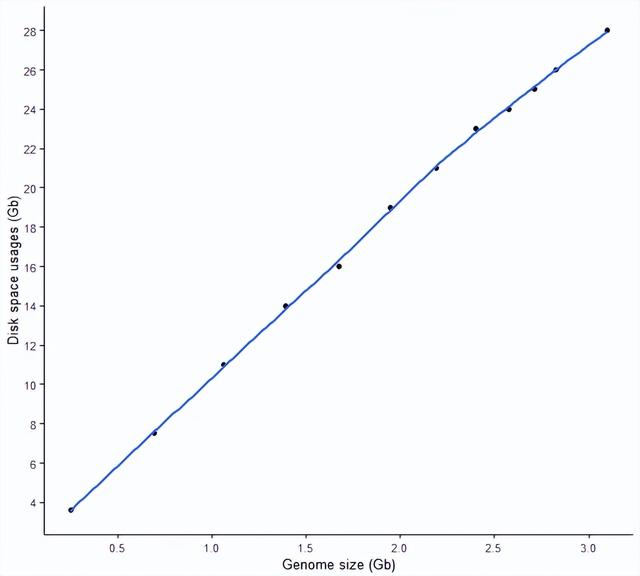
sp_scatterplot("GRCh38_star_genome_build.summary", melted = T, xvariable = "Data_size",
yvariable = "Index_size", smooth_method = "auto",
x_label ="Genome size (Gb)", y_label = "Disk space usages (Gb)") +
scale_x_continuous(breaks=seq(0.5,3, by=0.5)) +
scale_y_continuous(breaks=seq(2,28, by=2))